PJ Labs · macOS · lokale Genomik
Deine Genomdaten. Dein Mac. Ein professioneller Bericht.
Genome ist eine native Workbench für WGS-Daten: von FASTQ, BAM und CRAM über HLA, KIR, LPA, Pharmakogenetik, Repeat-Expansionen, SNP-Suche, Microarray-Exporte und Referenzverwaltung bis zu PDF-first Reports. Ohne Cloud und ohne Upload.
App-Rundgang
So fühlt es sich an, durch Genome zu klicken.
Wähle links einen App-Bereich. Das echte freigestellte Screenshot-Fenster wechselt wie beim Klick in der App.
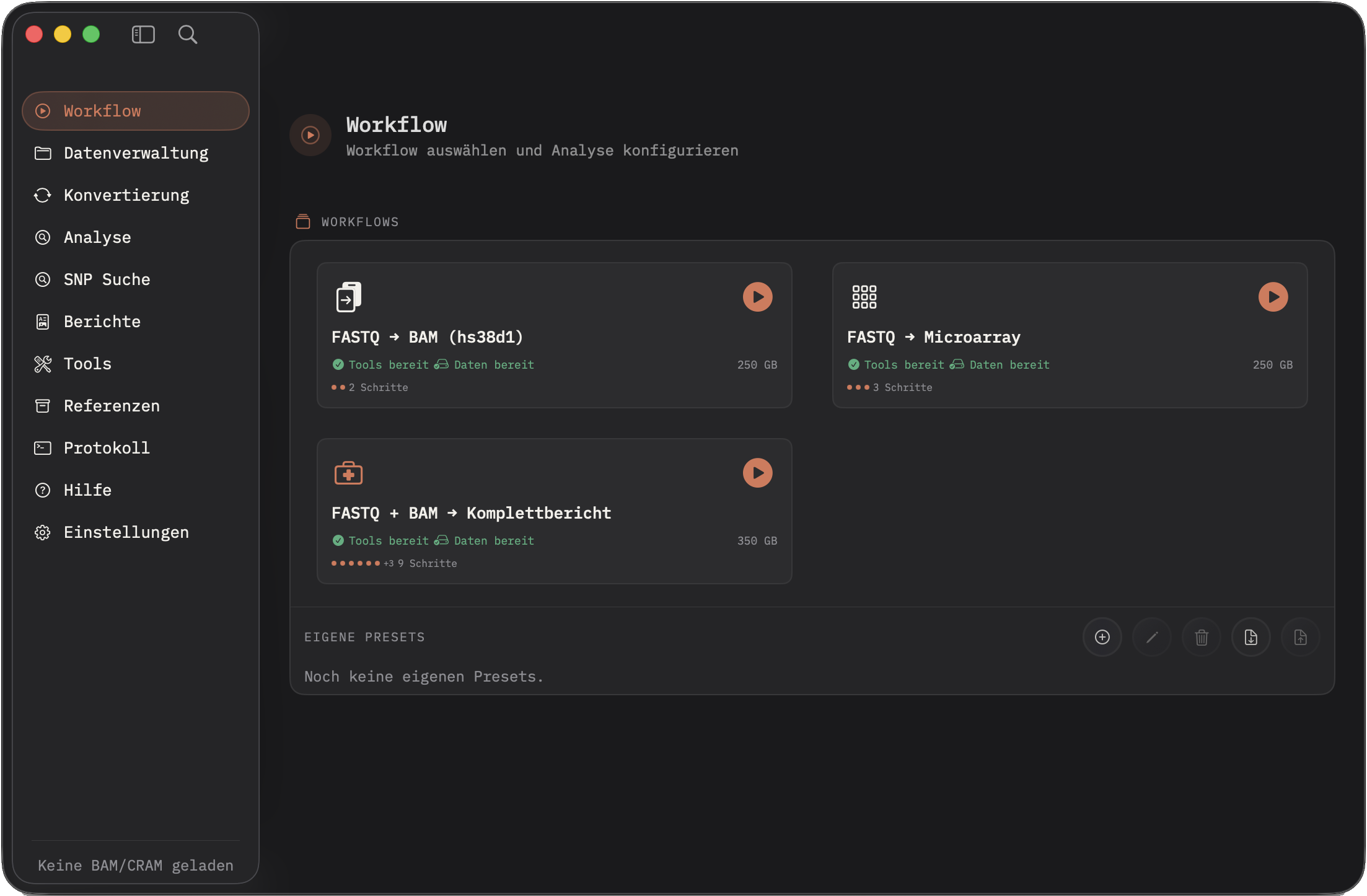
Workflow
Presets, Voraussetzungen, Start-Aktionen und eigene Workflows in einer ruhigen nativen Mac-Oberfläche.
Vollständigkeit
Genome ist nicht ein einzelner Report-Generator, sondern eine durchgehende lokale Genom-Workbench.
Workflow
Built-in- und Custom-Presets starten komplette Läufe: FASTQ→BAM, Microarray-Export, HLA/LPA/PGx/Repeat-Analyse und PDF-Berichte.
Datenverwaltung
BAM/CRAM laden, Index prüfen, Coverage/Chromosomenprofil lesen, Ordner öffnen, Pfade kopieren und Artefakte wiederfinden.
Konvertierung
FASTQ trimmen, alignen, sortieren, markieren, BAM↔CRAM konvertieren, VCF liften, Regionen extrahieren und Indizes erzeugen.
Analyse
Domänenspezifische Auswertungen statt Dashboard-Mix: mtDNA, EBV, HLA, KIR, Aldy, ExpansionHunter, KILDA, PGS, VCF und Microarray.
SNP-Suche
rsID-Listen bereinigen, in CombinedKit/23andMe/Ancestry-Daten suchen, Genotypen normalisieren und Coverage/Vertrauen anzeigen.
Berichte
Medizinische Genomik und Pharmakogenetik als primäre PDF-Ausgabe; HTML bleibt technischer Export.
Tools
Conda-, Homebrew- und Genome-managed Tools installieren, aktualisieren, deinstallieren, versionieren und validieren.
Referenzen
Referenzbibliothek, Bundles, HLA*LA-Graph, IPD-KIR/T1K-Ressourcen, Datenbankstatus und Tool-Smoke-Tests prüfen.
Protokoll & Hilfe
Operationen, Fehler, stille Phasen, Suchtreffer, Online-Hilfe und lokale Erklärungen bleiben auffindbar.
Workflows & Fachdomänen
Jede Funktion bleibt dort, wo sie fachlich hingehört, mit eigenen Inputs, Tool-Grenzen und Handoff in Berichte.
FASTQ → BAM/CRAM
fastp · bwa · samtools · mosdepth
Paired FASTQs werden getrimmt, mit bwa/bwa-mem2 oder minimap2 aligniert, sortiert, markdupliziert, indexiert und mit Coverage-/Flagstat-Checks protokolliert.
BAM/CRAM → Microarray
CombinedKit · 23andMe · Build 37/38
Genome erzeugt 23andMe, AncestryDNA, FTDNA, Living DNA, MyHeritage, GEDmatch und CombinedKit; Header und Build 37/38 bleiben konsistent.
Microarray Deep-Modus
CombinedKit · AD/DP · Konfidenz
Über den Genotyp hinaus schreibt Genome pro SNP die Allel-Tiefen (AD/DP), die Allel-Balance und eine Konfidenz (homozygot, heterozygot oder nicht beurteilbar) in die CombinedKit-Datei. Eine Kopfzeile fasst zusammen, wie viele Positionen sicher bestimmt sind.
HLA*LA + T1K
HLA*LA · T1K · IMGT/HLA
HLA*LA liefert G-Gruppen, Coverage, Q1/Q2 und DRB-Hinweise. T1K ergänzt HLA/KIR als zweite technische Evidenz und macht Konkordanz sichtbar.
KIR / T1K
KIR · IPD-KIR · T1K
KIR-Gene und KIR-HLA-Kontext werden getrennt vom HLA-Block geführt, mit IPD-KIR-Referenz, technischer Evidenz und klarer Gen-/Allel-Darstellung.
LPA / KILDA
KILDA · LPA · KIV-2
KILDA/KIV-2-Kontext wird als eigenes Modul behandelt. Rohwerte wie quantile=NA bleiben sichtbar statt als vermeintlich glatte Interpretation zu erscheinen.
Aldy / Pharmakogenetik
Aldy 4 · PharmCAT · CPIC/DPWG
Aldy 4 verarbeitet komplexe PGx-Gene; PharmCAT/SNP-Regeln ergänzen Variantenkontext. Aldy bleibt strikt Pharmakogenetik-only.
ExpansionHunter
Repeat · STR · ExpansionHunter
Repeat-Loci, Allelgrößen, Read-Evidenz und Konfidenzintervalle werden nach Upstream-Semantik gerendert, getrennt von SmallVariant-Genotypen.
mtDNA, Y, EBV & Microbiome
Haplogrep 3 · EBV · Kraken2
mtDNA- und Y-Artefakte, Haplogrep 3, EBV-Coverage/VCF sowie optionale Kraken2/Bracken-Kontexte bleiben als technische Evidenz erkennbar.
PGS & SNP-Kontext
PGS Catalog · rsID · ClinVar
PGS Catalog-Dateien, einzelne rsIDs, APOE/BRCA/HFE/F5/CYP-Beispiele und eigene SNP-Dateien werden lokal ausgewertet und als Kontext eingeordnet.
Toolchain
Genome wrappt Bioinformatik, versteckt sie aber nicht.
Die Tools-Seite zeigt Pfade, Versionen, Größen, Updates und Validierung. Installieren und deinstallieren umfasst die üblichen Alignment-Werkzeuge genauso wie KILDA, Aldy, ExpansionHunter, T1K, HLA*LA und Referenzen.
Core Alignment
fastp · fastqc · bwa/bwa-mem2 · minimap2 · samtools · sambamba · mosdepth · pigz
Variant & Utility
bcftools · tabix/bgzip · bedtools · Java · Picard · Python 3 · kallisto
Specialized Genomics
HLA*LA · T1K · KILDA · Nextflow · jellyfish · Aldy 4 · ExpansionHunter · Haplogrep 3
Optional Context
Kraken2 · Bracken · dbSNP · ClinVar · PGS Catalog · Genome+ Microarray Panels
Reports
PDF ist die primäre Ausgabe, mit sauberer Evidenz statt Marketing-Scores.
Medizinische Genomik
HLA, T1K-HLA/KIR, KILDA/LPA, Repeat-Expansionen, SNP-/ClinVar-Kontext, PGS, mtDNA/Haplogrep und technische Evidenz erscheinen nur, wenn passende Eingaben wirklich vorhanden sind.
Pharmakogenetik
PGx bleibt PGx: Aldy-Diplotypen, PharmCAT/SNP-Regeln, CPIC/DPWG/PharmGKB-Kontext und vorsichtige Sprache ohne Vermischung mit HLA/KIR-Rohanhängen.
Berichtsmodule
HLA & T1K-Konkordanz · KIR / T1K · Pharmakogenetik & Aldy · LPA / KILDA · Repeat-Expansionen · SNP/ClinVar/PGS-Kontext · mtDNA/Haplogrep · EBV & technische Evidenz
Lokal und nachvollziehbar
Keine Cloud, kein Account, kein Upload. Pfade, Versionen, Eingaben, Referenzen, Manifeste und Logs bleiben sichtbar.
Keine falsche Vereinfachung
Genome trennt Rohwert, Tool-Grenze, technische Evidenz, Kontext und vorsichtige Interpretation.
Apple-native Oberfläche
SwiftUI, Systemfarbe, Sidebar, kompakte Controls, PJDesignKit-Typografie und ruhige Karten statt Web-Dashboard.
Download
Genome auf deinem Mac ausprobieren.
Lokale Datenverarbeitung, strukturierte Hilfe, professionelle Berichte und eine Toolchain, die sichtbar bleibt.